- 产品详情
- 产品参数
- 产品评论
 | ——GSEA代码 |
GSEA全名为Gene Set Enrichment Analysis(基因集富集分析)。用以分析特定基因集(如关注的GO条目或KEGG Pathway)在两个生物学状态(如癌症与对照,高龄与低龄)中是否存在差异。能够研究基因变化的生物学意义。
普通GO/KEGG富集的思路是先筛选差异基因,然后确定这些差异基因的GO/KEGG注释,然后通过超几何分布计算出哪些通路富集到了,再通过p值或FDR等阈值进行筛选。挑选用于富集的基因有一定的主观性,没有关注到的基因的信息会被忽视,所以有一定的局限性。
在这种情况下有了GSEA(Gene Set Enrichment Analysis),其思路是发表于2005年的Gene set enrichment analysis: aknowledge-based approach for interpreting genome-wide expression profiles。主要是要有两个概念:预先定义的基因集S(基于先验知识的基因注释信息)和待分析基因集L(一般初始输入是表达矩阵);然后GSEA目的就是为了判断S基因集中的基因是随机分布于L(按差异表达程度对基因进行排序),还是聚集分布在L的顶部或者底部(也就是存在差异性富集)。如果基因集中的基因显著富集在L的顶部或者底部,这说明这些基因的表达对定义的分组(预先分组)的差异有显著影响(一致性)。在富集分析的理论中,GSEA可以认为是第二代,即Functional Class Scoring(FCS) Approaches。
基本原理
从方法上来讲,GSEA主要分为基因集进行排序、计算富集分数(Enrichment Score,ES)、估计富集分数的显著性水平并进行多重假设检验三个步骤。
第一步对输入的所有基因集L进行排序,通常来说初始输入的基因数据为表达矩阵,排序的过程相当于特定两组中(case-control、upper-lower等等)基因差异表达分析的过程。根据所有基因在两组样本的差异度量不同(共有六种差异度量,默认是signal 2 noise,GSEA官网有提供公式,也可以选择较为普遍的foldchange),对基因进行排序,并且Z-score标准化。
第二步是GSEA的核心步骤,通过分析预先定义基因集S在第一步获得的基因序列上的分布计算富集指数Enrichment Score,并绘制分布趋势图Enrichment plot。每个基因在基因集S的Enrichment Score取决于这个基因是否属于基因集S及其差异度量(如foldchange)。差异度量越大基因的Enrichment Score权重越大,如果基因在基因集S中则Enrichment Score取正,反则取负。将基因集L在基因集S里的所有基因的Enrichment Score一个个加起来,就是Enrichment plot上的Enrichment Score趋势,直到Enrichment Score达到最大值,就是基因集S最终的Enrichment Score。
第三步是为了检验第二部获得结果的统计学意义,对Enrichment Score进行标准化,计算标准化Enrichment Score(NES)。并计算显著性水平(NOM p-val)和矫正多重假设检验显著性水平(FDR q-val)。
GSEA术语解读
Enrichment score(ES)
ES是GSEA最初的结果,反应关注的基因集S在原始基因数据序列L的顶部或底部富集的程度。ES原理:扫描排序序列,当出现一个基因集S中的基因时,增加ES值,反之减少ES值,一个基因的ES值权重与差异表达度相关。ES是个动态值,最终ES是动态扫描过程中获得的最大ES值。如果最终ES为正,表示某一功能基因集S富集在排序序列顶部。ES为负,表示某一基因集S富集在排序序列底部。
NES
由于ES是根据分析的排序序列中的基因是否在一个基因集S中出现来计算的,但各个基因集S中包含的基因数目不同,且不同功能基因集S与原始数据之间的相关性也不同,因此比较数据中基因在不同基因集S中的富集程度要对ES进行标准化处理,也就是计算NES。
NES=某一基因集S的ES / 数据集所有随机组合得到的ES平均值,NES是主要的统计量。
nominal p-value(普通P值)
描述的是针对某一功能基因集S得到的富集得分的统计显著性,通常p越小富集性越好。
FDR(多重假设检验矫正P值)
NES确定后,需要判断其中可能包含的错误阳性发现率。FDR=25%意味着对此NES的判断4次可能错 1次。GSEA结果中,高亮显示FDR<25%的富集基因集S。因为从这些功能基因集S中最可能产生有意义的假设。大多数情况下,筛选FDR<25%是合适的,但是,假如分析的数据集样本较少,且选择的是探针随机组合而不是表型组合,则应该酌情选择更加严格的FDR阈值。一般而言,NES绝对值越大,FDR值就越小,说明富集程度高,结果更加可靠。
数据要求
1、通常为表达谱芯片或测序数据(已经过预处理),也可以是其他形式可排序的基因数据。
2、具有已知生物学意义(GO、Pathway、癌症特征基因集等)的基因集。
下游分析:
得到GSEA结果之后的分析有:
1. 基因注释
2. PPI基因相互作用分析与互作网络绘制
3. GSVA分析
图形示例:
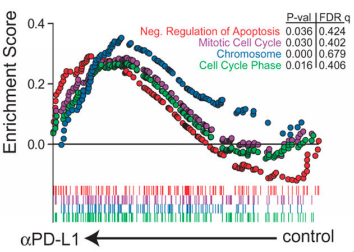
1、绘制基因集富集趋势图(Enrichment plot)
图注
横坐标:按差异表达差异排序的基因序列。数值越小(偏向左端)的基因代表在shICAM-1组中有越高倍数的差异表达,数值越小(偏向右端)的基因在对照组中有越高倍数的差异表达。
纵坐标:上方的纵坐标为富集打分ES,ES是一个动态的值,沿着基因序列,找到条目中的基因则增加评分,否则减少评分。通常用偏离0最远的值作为最终富集打分。下方的纵坐标代表基因表达与表型的关联,绝对值越大代表关联越强,数值大于0代表正相关,小于0则代表负相关。
2、绘制功能基因集S中基因在数据中的表达热图
图注:根据基因表达量作图,每个小方格表示特定基因在特定样本中的表达量,其颜色表示该基因表达量大小,颜色从红到蓝代表表达量从高到低。图片右侧标注基因信息,上方标注样本信息,色块分区代表样本分组。
应用示例:
文献1:A seven-miRNA expression-based prognosticsignature and its corresponding potential competing endogenous RNA network inearly pancreatic cancer.(于2019年7月发表在Exp Ther Med.,影响因子1.448)
早期胰腺癌中的基于7-miRNA表达量的预后预测模型及其ceRNA网络分析
为了研究挑选出的7-miRNA预后模型的生物学意义,作者通过GSEA分析查找受7-miRNA表达值影响的KEGG Pathway。并挑选了富集的KEGG Pathway中的基因进行ceRNA网络分析。
文献2:HSP90/AXL/eIF4E-regulated unfolded proteinresponse as an acquired vulnerability in drug-resistant KRAS-mutant lung cancer(于2019年8月发表在Oncogenesis.,影响因子5.995)
HSP90/AXL/eIF4E调控的未折叠蛋白反应作为KRAS突变的耐药性肺癌的突破口
作者使用GSEA对KRAS突变的癌症细胞系的基因表达数据进行了分析,分析了关键EMT信号通路的基因富集情况。
;){kind=link}