配对基因调控模式的模型构建
收藏
¥0.00
收藏
- 产品详情
- 产品参数
- 产品评论
研究目的:
用成对的基因gene-pair分析样本的特征,该特征后续可以和患者预后等其他特征关联分析。
应用场景:
1.当常规的差异分析不能找到有效结果时。
2.当只有患者的基因表达信息,没有正常对照时。
3. 微生物分析时,样本的微生物含量不高,表达矩阵稀疏时。
背景:
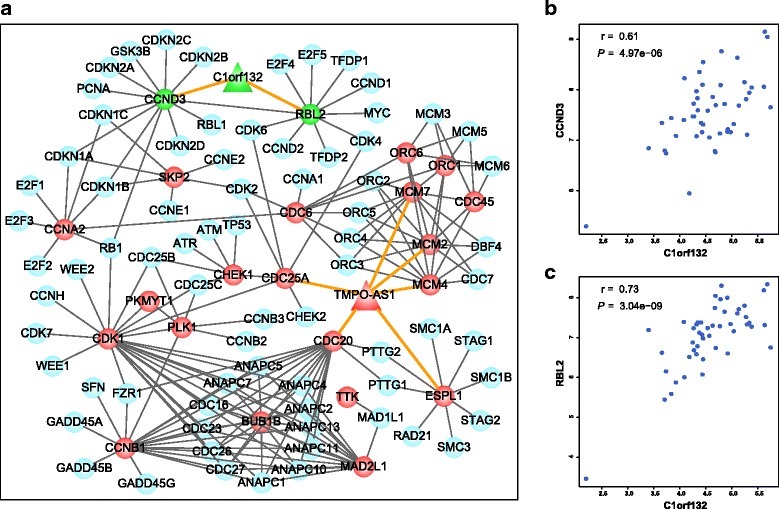
某些基因(如lncRNA、miRNA)在患者之间表达的高度异质性,传统的差异检验的方法不能很好的工作。研究者发现基因的相对表达排序在特定的正常人体组织中趋于高度稳定,而在相应的癌组织中则受到广泛干扰,基因表达水平之间的排序反向关系可用于识别个体患者中的 DE 基因。另外,基于排序-方法的优点,它对批次效应和数据归一化不敏感,因此可直接利用来自不同的数据集。这篇文章建立了一个新的算法,用成对的基因gene-pair构建预后模型。他们发现使用样本内的相对基因表达排名而不是绝对表达值得出的预后特征在来自不同实验室和平台的独立数据集中是可靠的。
例如A和B基因,A表达量 > B表达量设为1,A表达量 < B表达量设为0。筛选出基因互斥的配对,进行预后模型构建。获得的模型具有平台独立性,只要单样本基因之间表达具有可比性就可以了。

;){kind=link}