RNA-Seq前处理:mRNA富集还是rRNA去除?
除核糖体 RNA 外,样本还可能含有其他丰富的转录本,例如,血液样本中的珠蛋白 mRNA 可占所有 mRNA 分子的 30 – 80%。如果不除去这些占比很大的globin mRNA,我们测序所获得的大部分reads数将来自这些不需要的 RNA 种类,它们既占用了宝贵的测序空间并限制了测序的样本量。对于富集所需 RNA 或去除不需要的 RNA的方法,我们将在本章中讨论这些方法。

1. Poly(A) 富集
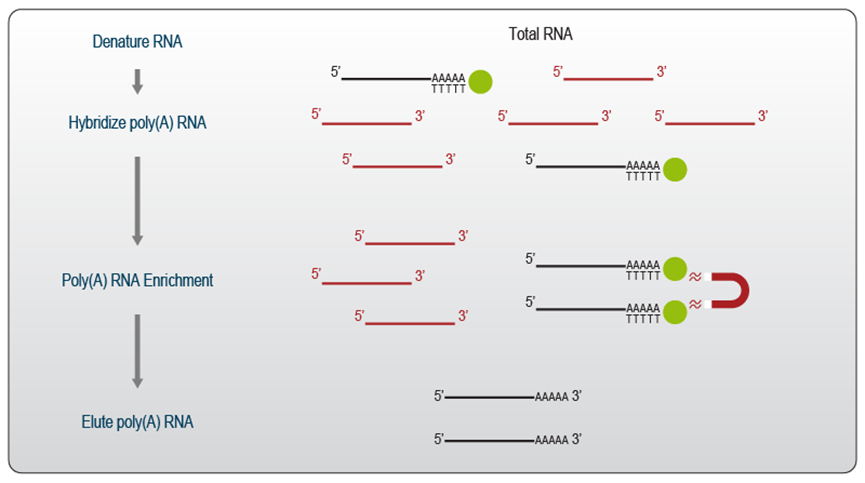
图1 | poly(A) RNA 分选示意图。
Poly(A) 富集是一种非常经济高效且快速的预处理方法,可以选择主要编码蛋白质的 mRNA。它可用于所有具有 poly(A) 尾 RNA 的物种(真核生物),以去除不需要的 rRNA 并将测序数据集中在 mRNA 上。
然而,poly(A) 富集有两个主要缺点:
➊首先,它只能用于具有poly(A)-尾RNAs的物种。因此,它仅限于真核生物,不能用于原核生物。然而,真核生物也拥有我们感兴趣的缺少 poly(A) 尾的转录本。它们在 poly(A) 选择过程中,也将与 rRNA 一起被移除。这些转录本包括 microRNA、小核仁 RNA (snoRNA)、转运 RNA (tRNA)、一些长非编码 RNA (lncRNA),甚至是蛋白质编码 mRNA,例如组蛋白 mRNA。因此,对这些类型的 RNA 或原核生物,研究人员通常利用 rRNA 去除而不是 poly(A) 选择。
➋其次,poly(A) 富集需要高质量的 RNA (RIN / RQN > 8)。降解导致转录本的断裂,并且由于对 poly(A) 尾的选择,3' 端被富集,而5' 序列则会丢失,导致对降解的 RNA 样本作为起始材料测序结果会产生强烈的 3' 偏好性。因此,rRNA耗尽或 3' mRNA-Seq 是处理降解 RNA 的首选方法。
2. 文库制备过程中的 Poly(A) 富集
也可以通过 oligo(dT) 反转录产生全长 cDNA。对于这些流程,逆转录反应针对长片段的生成进行了优化,并且 5' 端通常通过依赖帽的捕获方法或模板置换进行富集。
RNA-Seq 实验中不同的 DNA 拷贝数主要是由两个因素的引起的:
➊细胞中转录本的表达水平在几个数量级之间变化:一些转录本可以以每个细胞超过 10,000 个拷贝的方式存在,但其他转录本可能仅以非常低的水平表达,只有 1-2 个拷贝。
总 RNA 样本中最丰富的转录本是 rRNA、tRNA 和管家基因mRNA。此外,组织或样本特异性的过量转录本也属于这一类。
DSN处理如何去除高风度序列?
在 RNA-Seq 中,DSN 处理可用于部分均一化反映转录本动态范围的cDNA 浓度。这是通过去除高丰度转录本来实现的。DSN 处理通常在 cDNA 第一和第二链合成后进行。当然,当 RNA 模板尚未去除时,也可以在第一链合成后使用 DSN。
DSN 反应利用新合成的 cDNA 分子的杂交特性(图 2)。cDNA 合成后,在反应再次冷却之前,通过在高温下短暂孵育使分子变性。降低温度后,互补的 cDNA 链会重新退火(这个过程称为复性)。由于浓度更高,因此与互补链相互作用的机会更高,因此与低丰度 cDNA 相比,丰富的 cDNA 链重新退火更快、更有效。因此,大多数双链 cDNA 将来自丰富的转录本,而来自中等和低表达转录本的 cDNA 将保持单链。然后通过 DSN 切割双链 cDNA 部分,从而去除所有高丰度的序列,整个pool中的分子被均一化到相似的浓度水平。最后,剩余的 cDNA 分子在 PCR 反应中被扩增以生成可测序的文库(图 2)。
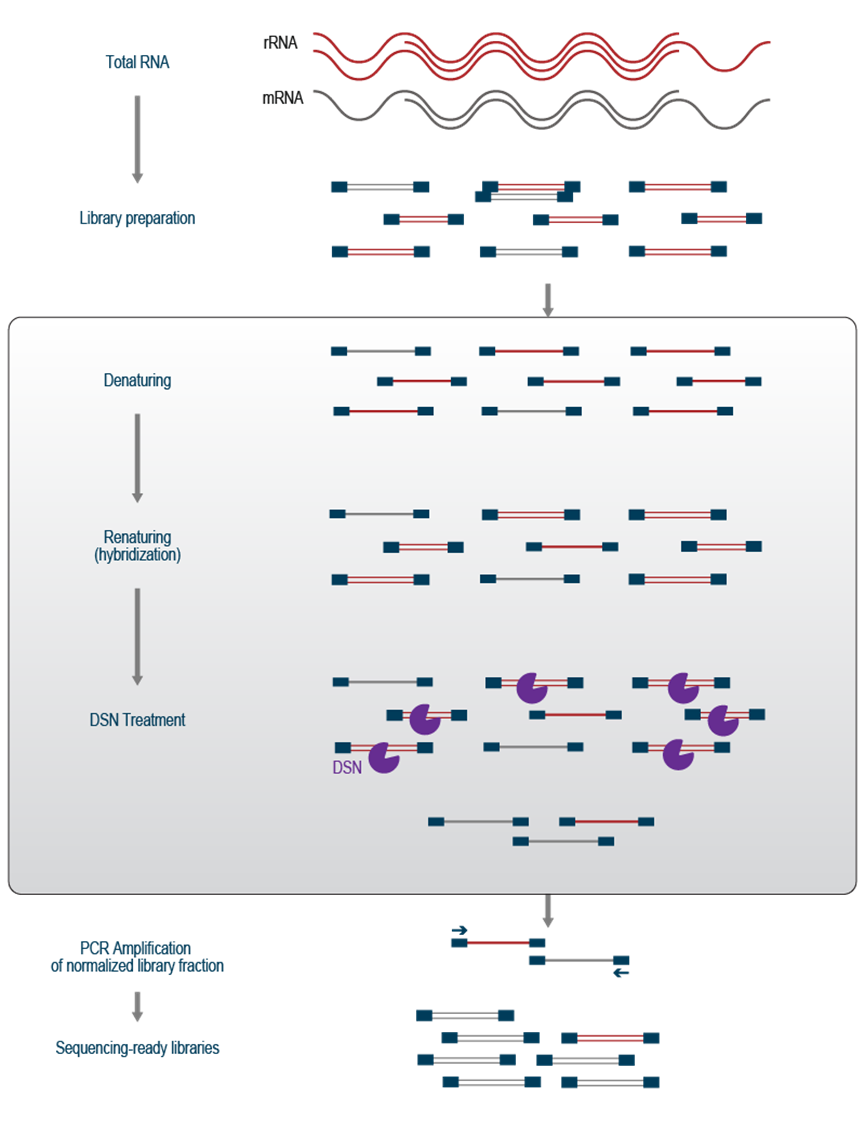
这种方法的主要缺点是去除是非特异性的,并且针对任何高丰度的序列。如果您感兴趣的转录本属于较高拷贝数类别,则它也可能会受到 DSN 介导的降解。此外,当需要定量信息来评估转录本表达水平的变化时,应特别慎用。根据使用的input量,DSN 处理可能会使浓度均一化,从而无法正确量化表达水平的变化。
4. 基于探针的去除技术
杂交/捕获技术
基于杂交/捕获的去除方法使用一组专门针对 rRNA 序列的亲和探针。探针的数量和位置取决于目标物种的数量、目标组中核糖体 RNA 序列的复杂性以及靶向降解 RNA 的兼容性。为了有效去除降解样品中的 rRNA,目标序列上的探针密度需要更高,因为目标区域的断裂会损害探针在高温下的杂交。探针包含亲和标签,允许使用具有相应结合位点的磁珠进行捕获。因此,探针混合物中包含的探针数量与用于捕获的珠子的结合能力密切相关。增加探针分子的数量,例如,通过使用非常高密度的探针或针对一大群不同的物种为靶标的探针,可能会对去除效率产生反作用,因为它可能会导致磁珠超载。为确保最佳结果,杂交/捕获方法的探针和捕获磁珠应以最佳比例滴定
在第一步中,亲和探针与总 RNA 混合并变性,从而促进探针接近高度结构化的目标序列。杂交在高温下进行,以确保特异性结合并最大限度地减少不希望的脱靶去除效应。捕获磁珠用于从溶液中去除与核糖体 RNA 杂交的探针。最后的纯化步骤去除所有反应成分并回收剩余的 RNA 用于下游应用(图 3)。启衡星生物可以提供任意物种的rRNA去除试剂盒!
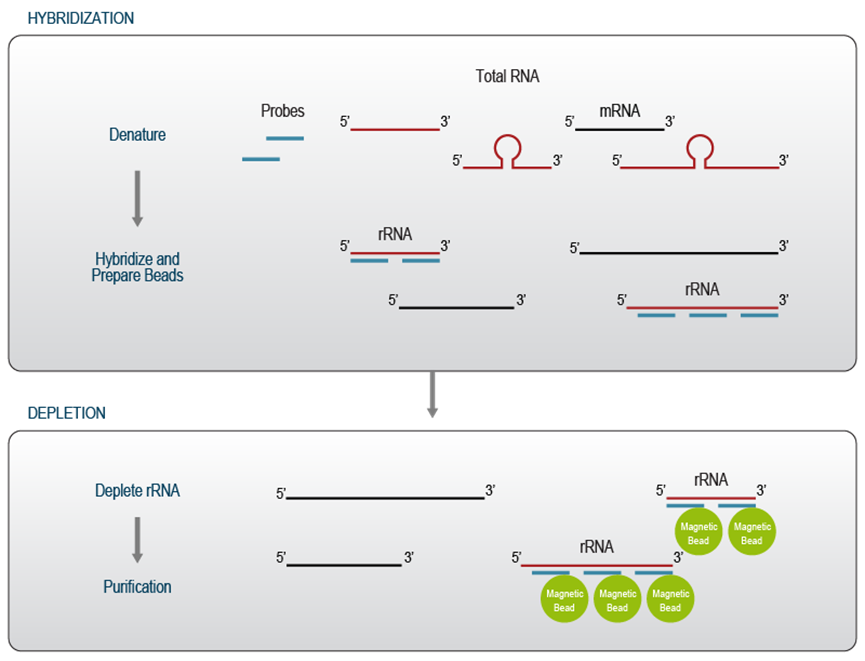
杂交/捕获方法不依赖于酶促反应,因此这些方法保留完整的全长转录本以供下游应用。它们特别适用于具有挑战性的应用,例如 RiboSeq 4,并最大限度地减少非特异性 RNA 降解。
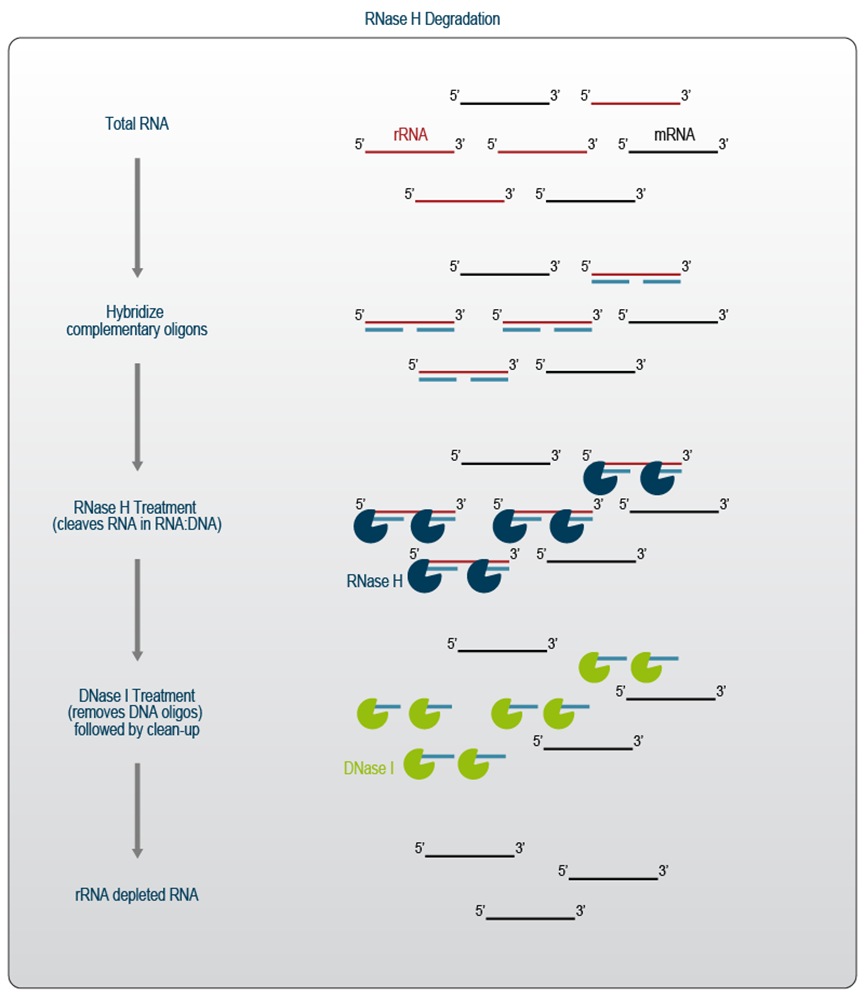
基于RNase H的降解被广泛使用,因为反应组分和寡核苷酸价格便宜,而且它们的探针数量可以增加以覆盖许多物种。然而,该方法基于降解的性质带来了非特异性去除珍贵转录本的风险,也使其不适用于某些应用。例如,依赖于在耗尽之前添加 DNA 接头的 RNA-Seq 工作流程不应进行RNase H / DNase I 处理,因为这也会降解 RNA-DNA 融合分子。最近的研究结果表明,基于RNase H的方法可能不适合具有挑战性的应用,例如 RiboSeq 4。
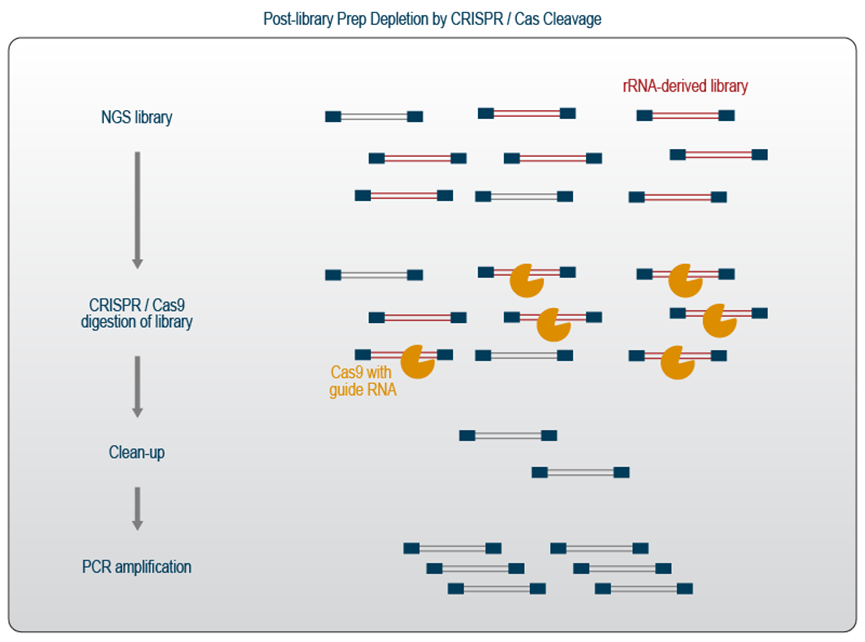
文库制备后基于CRISPR-Cas9的去除
该系统可以通过提供特定的导向RNA来靶向任何想要的序列来开发。在RNA-Seq方法中,CRISPR-Cas9与靶向rRNA序列或其他丰富序列的引导RNA一起使用,在文库制备完成后可以方便地对rRNA或其他高丰度转录本的文库片段进行降解。
准备好测序的文库与 Cas9 核酸酶一起孵育,该核酸酶已与特定的 rRNA 向导预复合(图 5)。然后在分子库中切割包含任何目标序列的所有文库。孵育之后是清洗步骤,以去除短的文库片段和被切割的片段。由于此过程去除了大部分文库片段,剩余的分子将通过另一轮 PCR 重新扩增。
参考文献:
;){kind=link}